Subject archive for "lime"

SHAP and LIME Python Libraries: Part 2 - Using SHAP and LIME
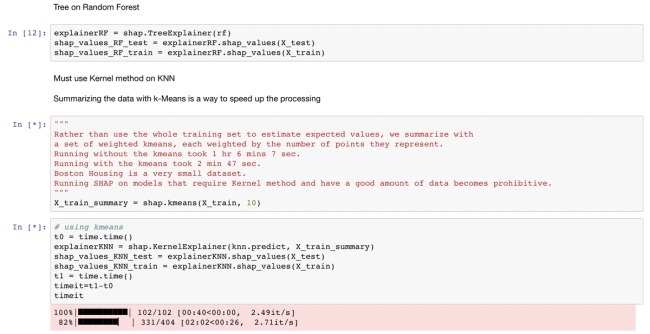
This blog post provides insights on how to use the SHAP and LIME Python libraries in practice and how to interpret their output, helping readers prepare to produce model explanations in their own work. If interested in a visual walk-through of this post, then consider attending the webinar.
By Josh Poduska9 min read
SHAP and LIME Python Libraries: Part 1 - Great Explainers, with Pros and Cons to Both
This blog post provides a brief technical introduction to the SHAP and LIME Python libraries, followed by code and output to highlight a few pros and cons of each. If interested in a visual walk-through of this post, consider attending the webinar.
By Josh Poduska6 min read

Trust in LIME: Yes, No, Maybe So?
In this Domino Data Science Field Note, we briefly discuss an algorithm and framework for generating explanations, LIME (Local Interpretable Model-Agnostic Explanations), that may help data scientists, machine learning researchers, and engineers decide whether to trust the predictions of any classifier in any model, including seemingly “black box” models.
By Ann Spencer7 min read
Subscribe to the Domino Newsletter
Receive data science tips and tutorials from leading Data Science leaders, right to your inbox.
By submitting this form you agree to receive communications from Domino related to products and services in accordance with Domino's privacy policy and may opt-out at anytime.